Heute kennen wir unter dem Begriff vier verschiedene genetische Formen der Hämochromatose, die als
Typ1 bis Typ 4 bezeichnet werden (6):
Typ1 HFE-Protein mutiert. Häufigste Form in Nordeuropa, USA, Australien (1 : 200-300); autosomal
rezessiv; parenchymale Eisenspeicherung: variabler, häufig milder Phänotyp (Leber, Gelenke, endokrine
Organe); spricht gut auf Aderlass an.
Typ 2A HJV-Protein mutiert
Typ 2B HAMP-Gen betroffen. Beides seltene Formen, Juvenile Hämochromatose, autosomal rezessiv; früh
einsetzende Eisenüberladung: häufig schwere Organschäden (Leber, endokrine Organe); spricht gut auf
Aderlass an.
Typ 3 TfR2-Protein betroffen. Sehr ähnlich Typ1. Häufiger in Südeuropa, autosomal rezessiv;
parenchymale Eisenspeicherung: variabler, häufig milder Phänotyp (Leber, Gelenke, endokrine Organe);
spricht gut auf Aderlass an.
Typ 4 (A und B) Ferroportin-Gen betroffen. Selten. Autosomal dominant; retikuloendotheliale
Eisenspeicherung: meist geringe Organbeteiligung, spricht teilweise schlecht auf Aderlass an.
Diese unterschiedlichen Formen haben alle was mit der Synthese von Hepcidin in der Leber (Typ 1-3) oder
der Wirkung von Hepcidin auf den Eisenexport von Zellen (Enterozyten im Darm, Makrophagen) zu Tun
(Abb. 1)
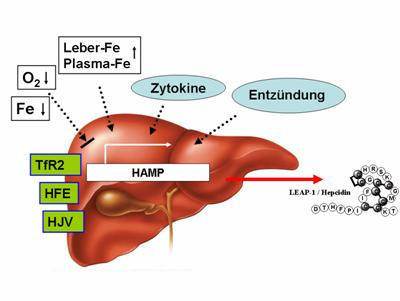
Abb. 1: Regulation der Hepcidinsynthese (HAMP-Gen) in der Leber. Mutationen im HFE-, TfR2- und HJV-
Gen (Typ 1-3 Hämochromatose) beeinträchtigen dabei direkt die Biosynthese von Hepcidin in der Leber.
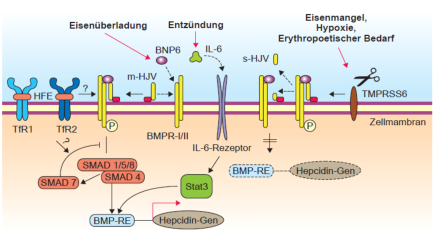
Abb. 2: Signaltransduktion der Hepcidinsynthese in der Leber. Das HFE-Protein, Transferrin-Rezeptor 2
und Hämojulelin (HJV) sind daran beteiligt. Auch andere Stimuli wie Entzündung führen zu einer
Hepcinsynthese.
HFE-assoziierte Hämochromatose; Typ 1 Hämochromatose
Diese Form der hereditären Hämochromatose ist eine der häufigsten monogen vererbten Krankheiten der
kaukasischen Bevölkerung Nordeuropas, Amerikas, Australiens. Die Homozygoten-Frequenz ist ungefähr 1
auf 300 Personen in der Normalbevölkerung. Die Gen-Frequenz ist 1:20, die Heterozygotenhäufigkeit ist 1
auf 10 Normalpersonen (6). Die Genhäufigkeit legt nahe, dass die heterozygote Form einen
Selektionsvorteil in der Evolution geboten hat. Heterozygote Genträger haben gegenüber der
Normbevölkerung eine leicht positive Eisenbilanz, was während ausgedehnter Hungerphasen, bei häufigen
Schwangerschaften oder für eine verbesserte Abwehr gegen Infektionen hilfreich gewesen sein kann.
Molekularbiologische Grundlagen
In einer sehr aufwendigen Suchaktion wurde 1996 von der amerikanischen Firma Mercator Genetics, die
eigens zu diesem Zweck gegründet wurde, das wahrscheinliche Hämochromatose-Gen (HFE) mittels
„positional cloning" lokalisiert und die für die Krankheit ursächliche Punktmutation identifiziert (Feder et al.
1996, 1997, 1998).
In dem HFE-Gen von Patienten mit Hämochromatose wurde eine Punktmutation gefunden, bei der eine
Transition von Guanin 845 nach Adenin vorliegt. Dies führt im korrespondierenden Polypeptid zu einem
Aminosäureaustausch Cystein 282 nach Tyrosin (Cys282Tyr bzw. C282Y). Die Primärstruktur des
zugehörigen Proteins ähnelt sehr dem Antigen-präsentierenden HLA-A-Protein, zeigt aber nicht dessen
Polymorphismus. Von der Primärstruktur kann abgeleitet werden, dass das HFE-Protein ß2-Mikroglobulin
nichtkovalent bindet. Offenbar wird durch diese Mutation die Bindung von ß2-Mikroglobulin an das HFE-
Genprodukt blockiert (Feder et al. 1996, Lebron et al. 1998). Dies passt sehr gut zu der Beobachtung, daß
für ß2-Mikroglobulin defiziente Mäuse spontan eine hämochromatose-ähnliche Eisenüberladung entwickeln
(DeSousa et al. 1994, Rothenberg und Voland 1996, Santos et al. 1997). Inzwischen gibt es auch eine
HFE-Gen „knockout"-Maus, die bezüglich gesteigerter intestinaler Eisenaufnahme und parenchymale
Eisenüberladung genau der hereditären Hämochromatose beim Menschen entspricht (Zhou et al. 1998).
Der erste Hinweis auf die direkte Verknüpfung des HFE Gen mit dem Eisenstoffwechsel ergab sich durch
die Beobachtung, daß das HFE-Protein mit dem Transferrin Rezeptor einen Komplex bildet und offenbar die
Affinität des TfR zu Transferrin herabsetzt. Das C282Y-mutierte Protein zeigt diese Bindung und Wirkung
nicht (Feder et al. 1996, Lebron et al. 1998).
Eine zweite Mutation im HFE-Gen, Histidin 63 =>Asparaginsäure (H63D) kommt zu ca. 15 % in der
Normalbevölkerung vor, offenbar aber nicht zusammen mit der C282Y Mutation in einem Allel (Feder et al.
1996). Bei Personen mit klinischem Verdacht auf hereditäre Hämochromatose findet man in einigen Studien
signifikant häufiger heterozygote Gen-Träger für die C282Y-Mutation die auch heterozygot für die H63D
Mutation sind. Möglicherweise führt diese „Compound"-Heterozygotie ebenfalls zu einer klinisch relevanten
Eisenüberladung (Feder et al. 1996). Es bleibt abzuwarten, ob die H63D-Mutation im Rahmen der erblichen
Eisenspeicherkrankheit bzw. bei Lebererkrankungen mit evtl. begleitender leichter Eisenüberladung eine
pathophysiologische Relevanz besitzt und damit auch langfristig eine diagnostische Bedeutung gewinnen
wird.
Das HFE-Gen wird in allen Geweben exprimiert (Leber, Duodenum, Herz, Pankreas), die bei
Hämochromatose eine Rolle spielen (Feder et al. 1996).
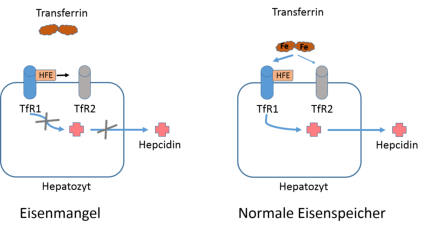
Abb. 3: Model der Hepcidinsynthese in Hepatozyten. Hepatozyten haben den Transferrinrezeptor 1 (TfR1) und 2 (TfR2)
exprimiert, die sich an der Obefläche befinden. TfR1 hat eine wesentlich höhere Affinität zu Transferrin als TfR2 und
beide können das HFE-Protein binden. Bei Eisenmangel bindet kein Transferrin an TfR1 und das HFE-Protein wird an
TfR2 abgegeben. In dieser Situation wird physiologisch kein Hecidin gebildet und die intestinale Eisenabsorption bleibt
hochreguliert. Bei hohem Serum-Eisen bindet Transferrin vorallem an TfR1 und es wird im Hepatocyten Hepcidin
gebildet. Model nach Montalbetti et al. 2013.
Genetik
Die hereditäre Hämochromatose eine häufige, wenn nicht die häufigste, genetisch bedingte Krankheit der
kaukasischen Bevölkerung Nordeuropas, Amerikas, Australiens. Die Homozygoten-Frequenz (q2) ist
ungefähr 1 auf 400 Personen in der Normalbevölkerung. Die Gen-Frequenz (q) ist 1:20, die
Heterozygotenhäufigkeit (2pq, Hardy-Weinberg- Gleichgewicht) ist 1 auf 10 Normalpersonen (Powell 1994).
Die Häufigkeit der erblichen Eisenspeicherkrankheit ist damit weit größer als die der Phenylketonurie, der
zystischen Fibrose und der Muskeldystrophie zusammengenommen. Die Inzidenz der Hämochromatose ist
z.B. in den USA größer als das Auftreten von AIDS. Die Genhäufigkeit legt nahe, dass die heterozygote
Form einen Selektionsvorteil in der Evolution geboten haben könnte. Denkbar wäre, dass eine leicht
positive Eisenbilanz bei Heterozygoten hilfreich gewesen sein mag während ausgedehnter Hungerphasen,
bei häufigen Schwangerschaften oder bei verbesserter Abwehr gegen Infektionen.
In nicht-kaukasischen Bevölkerungen (Ur-Australier, Chinesen) ist die Häufigkeit deutlich geringer (Gen-
Frequenz 0.38 %) (Cullen et al. 1998). In diesen Völkern ist die Hämochromatose-Mutation wahrscheinlich
durch Vermischung mit Kaukasiern schon in früher Zeit eingewandert.
Durch Studien von Marcel Simon in Frankreich wurde deutlich, dass es sich bei der hereditären
Hämochromatose um eine autosomal rezessiv vererbte Krankheit handelt (Simon et al. 1977). Dies ergab
sich aus der Tatsache, dass eine starke Assoziation der Krankheit mit dem HLA-A3 Antigen auf Chromosom
6 besteht. 75 % der Hämochromatose-Patienten einer Serie waren A3 positiv gegenüber 25 % in der
Kontrollgruppe. Innerhalb von Familien konnte durch die HLA-Typisierung (A und B-Lokus) die Vererbung
des Hämochromatose-Gens genau verfolgt werden. Weitere Studien führten zu der Annahme, daß es sich
bei der Hämochromatose um eine singulär aufgetretene Veränderung der DNA handeln muß. Das
Ursprungs-Hämochromatose-Gen entstand wahrscheinlich in der Steinzeit auf einem Chromosom mit dem
zufälligen HLA-Haplotyp A3 B7.
Durch seltene Rekombinationsereignisse ergibt sich heute bei einem Teil der Patienten die Situation, dass
das Hämochromatose-Allel nicht mit A3 assoziiert ist, sondern mit anderen A-Antigenen. Deshalb ist die
HLA-Typisierung in der Normalbevölkerung keine geeignete Screening-Methode auf die Hämochromatose.
Man hat sogar versucht, den Ur-Patienten lokal einzugrenzen. Die wahrscheinlichste Annahme geht von
einem keltischen Vorfahren in der Bretagne aus, und die Verbreitung der Krankheit in der kaukasischen
Bevölkerung folgt der Keltenwanderung (Simon et al. 1980, Mercier et al. 1998).
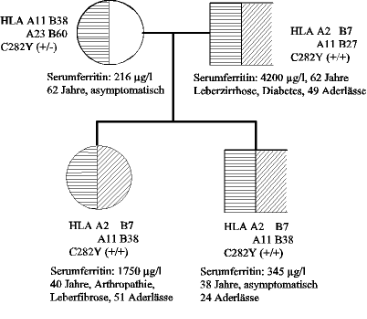
Abb. 4: zeigt die autosomal rezessive Vererbung in einem typischen Fall, in dem beide Elternteile des
Patienten heterozygote Genträger sind. Für jedes der Geschwister resultiert nach Mendelschem Erbgesetz
eine Wahrscheinlichkeit von 25 %, ebenfalls homozygot betroffen zu sein. Mit 25 %iger Wahrscheinlichkeit
ist ein Geschwisterteil reinerbig, zu 50 % ein heterozygoter Genträger.
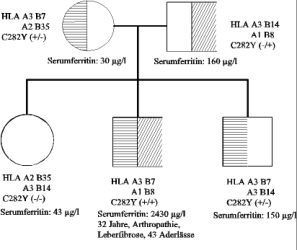
Abb. 5: „Pseudo-"autosomal dominante Vererbung der hereditären Hämochromatose in einer Familie, in der
ein Elternteil Genträger (wie üblich) das andere Elternteil aber selbst homozygot betroffen ist. Für jedes
Kind resultiert in diesem Fall eine Chance von 50 % homozygot (wie in dieser Familie beide Kinder)
betroffen zu sein (Alter, Laborwerte jeweils bei Diagnosestellung)
Die für Familienuntersuchungen bis vor kurzer Zeit so wertvolle HLA-Typisierung wird zukünftig zugunsten
der direkten HFE-Mutationsdiagnostik (C282Y-Mutation) aufgegeben werden.
Ein Beispiel einer „pseudo-autosomal" dominanten Vererbung zeigt die Familie in Abb. 5. Hier sind der
Vater und beide Kinder in homozygoter Form betroffen. Diese Konstellation ist möglich, wenn die Mutter
auch zufällig Genträger für die Hämochromatose ist, so dass in der Elterngeneration 3 von 4 Chromosomen
mutiert sind. Bei der Heterozygotenhäufigkeit von ca. 1:10 in der Normalbevölkerung ist diese Möglichkeit
nicht einmal so unwahrscheinlich, so dass eine entsprechende Familienuntersuchung nicht nur auf die
leiblichen Geschwister, sondern auch auf Kinder bzw. Eltern auszurichten ist.
Molekulare Diagnostik
Mit der Klonierung des HFE-Gens und der Identifizierung der zwei wichtigen Mutationen (C282Y und H63D)
sind alle Informationen zur Durchführung einer molekularen Diagnostik allgemein verfügbar. Die cDNA von
HFE ist in der Gensequenz-Datenbank (Genbank) des National Institute of Health (NIH) unter der Nummer
U60319 abgelegt und über die Adresse des National Center for Biotechnology Information
Das Testsystem von Feder et al. ist evtl. in leicht vereinfachter Form von verschiedenen Autoren auch aus
Deutschland verwendet worden (Arnold et al. 1998, Gottschalk et al. 1998). Nach der DNA-Isolierung aus
Vollblut wird der Bereich um die betreffende Mutation durch Verwendung von spezifischen Primern mittels
Polymerase-Kettenreaktion (PCR) amplifiziert und durch Restriktionsananalyse charakterisiert. Dabei
müssen nicht unbedingt die in der Originalarbeit verwendeten Primer benutzt werden. Wir haben in
unserem Labor seit April 1997 eine Methode im Einsatz, die auch in Labors einer portugiesischen (Porto et
al. 1998) und schwedischen Arbeitsgruppe (Cardoso et al. 1998) verwendet wird (Abb. 6). Inzwischen gibt
es kommerzielle Anbieter auf dem Deutschen Markt (z.B.Hain Diagnostika, Nehren), die eine PCR-
Methode mit sogen. reverser Hybridisierung anbietet, bei der amplifizierte DNA des Patienten an
spezifische Gensonden bindet, die an Nitrocellulosestreifen immobilisiert sind.
Prävalenz der HFE-Mutationen in einem Kollektiv von Hämochromatose-Patienten und Kontrollen aus
Norddeutschland (Nielsen et al. 1998a,b)
Es wurde ein eigenes Kollektiv von unverwandten Patienten (n=104) aus dem Norddeutschen Raum
untersucht, die vom klinischen, biochemischen und histopathologischen Standpunkt als homozygot
eingestuft waren.
Homozygotie für die erbliche Eisenspeicherkrankheit wurde angenommen, wenn mindestens drei der
folgenden Kriterien erfüllt waren:
a.) Serum-Ferritin > 300 µg/l ; b.) Transferrin-Fe-Sättigung > 62 %.; c.) Leber- Eisenkonzentration größer
1500 µg/g Leber, d.) hepatischer Eisenindex (Leber-Fe- Konz./Lebensalter) > 24 µg/g/Jahr, e.) mehr als 4 g
Speichereisen mobilisierbar durch Aderlasstherapie.
Ausgehend von jedem Patienten wurde eine Familienuntersuchung der Verwandten 1. Grades angestrebt.
Dabei wurden weitere 42 Personen auf die HFE-Mutationen analysiert. Zusätzlich wurden 157 erwachsene
Kontrollpersonen aus dem Norddeutschen Raum untersucht ).
Patienten
In der Patientengruppe waren 94.2 % homozygot und 5,8 % heterozygot für die C282Y Mutation. Bezüglich
Eisenbeladung war kein Unterschied zwischen diesen beiden Untergruppen erkennbar. In 196
Chromosomen der homozygoten Patienten wurde keine H63D Mutation gefunden. 4 von 6 Chromosomen
der heterozygoten Patienten, die keine C282Y-Mutation aufwiesen (=H63D-Risikochromosomen), wiesen
dagegen auch die H63D Mutation auf.
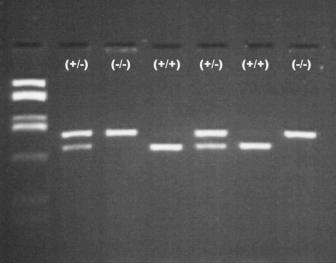
Abb.6 Typisches Bild einer PCR-Analyse mit Restriktionsenzymbehandlung auf die C282Y-Mutation. 2 und
4. Bande von links: heterozygote Genträger mit einer C282Y-Mutation (50 % der DNA wird geschnitten).
Bande 3 und 7 Wildtyp (keine Reaktion). von links, Bande 4 und 6, homozygote Träger der C282Y-Mutation
Familienmitglieder
In dieser Gruppe fanden sich 14 Personen mit homozygoter und 16 mit heterozygoter C282Y Mutation. 6
von 35 Risiko-Chromosomen trugen die H63D Mutation, darunter waren 3 Personen mit kombinierter
Heterozygotie. Im Vergleich mit heterozygoten C282Y-Trägern aus der Kontrollgruppe zeigten sich keinerlei
Unterschiede in den Werten für Transferrin-Fe-Sättigung und Serum-Ferritin.
Kontrollgruppe
Es wurde keine Personen mit homozygoter C282Y Mutation gefunden. 15 von 314 Chromosomen trugen
die C282Y-Mutation (Gen-Frequenz: 0.048). 40 von 299 Risiko-Chromosomen wiesen die H63D Mutation
auf (Gen-Frequenz: 0.13). Zwei Personen (1.3 %) waren homozygot, 23.6 % heterozygot für die H63D-
Mutation.
Die große Häufigkeit der C282Y-Mutation in homozygoter Form in norddeutschen Patienten stimmt gut mit
den Ergebnissen aus anderen Untersuchungen von kaukasischen Bevölkerungen überein (Feder et al.
1996, Merryweather-Clarke et al. 1997, Jazwinska et al. 1996, Jouanolle et al. 1997, Cardoso et al. 1998).
Eine aktuelle Studie aus Frankfurt kommt zu einer vergleichbar hohen Zahl von C282Y-Homozygoten in
Deutschland (Gottschalk et al. 1998). In nicht-kaukasischer Bevölkerungen (Ur-Australier, Chinesen) ist die
Häufigkeit sehr viel geringer (Allel-Frequenz 0.38 %) (Cullen et al. 1998) und die HFE-Mutationen sind
wahrscheinlich durch Vermischung mit Kaukasiern schon in früher Zeit eingewandert.
Die große Häufigkeit der C282Y-Mutation in homozygoter Form in norddeutschen Patienten stimmt gut mit
den Ergebnissen aus anderen Untersuchungen von kaukasischen Bevölkerungen überein (Feder et al.
1996, Merryweather-Clarke et al. 1997, Jazwinska et al. 1996, Jouanolle et al. 1997, Cardoso et al. 1998).
Eine Studie aus Frankfurt kommt zu einer vergleichbar hohen Zahl von C282Y-Homozygoten in
Deutschland (Gottschalk et al. 1998). In nicht-kaukasischer Bevölkerungen (Ur-Australier, Chinesen) ist die
Häufigkeit sehr viel geringer (Allel-Frequenz 0.38 %) (Cullen et al. 1998) und die HFE-Mutationen sind
wahrscheinlich durch Vermischung mit Kaukasiern schon in früher Zeit eingewandert.
In Südeuropäischen Ländern wie Frankreich (nur bestimmte Regionen!), Italien und Griechenland scheint
die Prävalenz der C282Y-Mutation ebenfalls deutlich niedriger zu liegen (50-70 %) (Borot et al. 1997,
Carella et al. 1997, Piperno et al. 1998). Dies könnte auf eine weitere Mutation hindeuten, die zu
Eisenüberladung führt und nur in südeuropäischen Bevölkerungen vorkommt.
Ähnlich wie in anderen Studien wurde eine hohe Zahl von kombinierten-Heterozygoten (C282Y/H63D) in
der Gruppe der eisenüberladenen Patienten gefunden (Feder et al. 1996). Die Ursache für die auch hier
wieder bestätigte komplette „linkage disequilibrium" zwischen der C282Y und der H63D Mutation ist weiter
unklar.
In der Kontrollgruppe von 157 unverwandten Personen fand sich kein Fall von Eisenüberladung und keine
homozygote C282Y Mutation. 15 von 314 Chromosomen wiesen die C282Y Mutation auf, was einer Allel-
Frequenz von 4.8 % entspricht. Unterstellt man, dass diese Mutation die einzig notwendige Ursache für
hereditäre Hämochromatose ist, dann kann man aus dieser Zahl eine Prävalenz für homozygote
Hämochromatose von 1: 440 errechnen. Dies ist in sehr guter Übereinstimmung mit einer von uns
durchgeführten Screening-Studie in 2812 prospektiven Blutspendern in Hamburg, bei der 7 Probanden mit
Eisenüberladung gefunden wurden (Prävalenz 1:402) (Nielsen et al. 1995a).
Vergleicht man die Werte für Transferrin-Sättigung und Serum-Ferritin zwischen heterozygoten Trägern der
C282Y-Mutation aus der Familien- und der Kontrollgruppe, so ergeben sich keine signifikanten
Unterschiede.
Dies zeigt an, dass es offenbar keine Unterschiede zwischen den „erprobten" Hämochromatose-Genen aus
der Familiengruppe und den „Zufalls"-Genen aus der Kontrollgruppe gibt. Dies ist ein Argument für die
These, daß die C282Y-Mutation die einzige und hinreichend notwendige Ursache für die Hämochromatose
ist. In Übereinstimmung mit anderen Untersuchungen zeigte sich in unserer Studie, dass es einige
eisenüberladene Patienten gibt, die nicht homozygot für die C282Y-Mutation sind.
Bei der Abklärung von Probanden mit V.a. Eisenüberladung fanden sich insgesamt auch 12Probanden mit
homozygoter H63D-Mutation. In allen diesen Fällen waren in der Vorgeschichte erhöhte Blutwerte (Serum-
Eisen oder Serum-Ferritin) bekannt, was bei unserer Untersuchung teilweise bestätigt wurde.
In zwei der Fälle zeigte sich eine leichtgradige Eisenüberladung, die eingangs definierten Kriterien für eine
hereditäre Hämochromatose waren aber nicht erfüllt. In der Literatur finden sich zwei Abstracts mit 4 bzw. 6
H63D-homozygoten Patienten, die mittelgradig eisenüberladen waren (Messerschmitt et al. 1998, Sham et
al. 1998). Möglicherweise führt also auch diese Konstellation in Einzelfällen zu einer relevanten
Eisenüberladung. Dies liefert ein weiteres Argument dafür, die H63D-Mutation in Verdachtsfällen auf hered.
Hämochromatose immer mitzubestimmen.
Andererseits gibt es auch Patienten mit homozygoter C282Y-Mutation, die nur wenig oder nicht
eisenüberladen sind. Diese Ausnahmen sprechen dafür, dass nichtgenetische Faktoren, wie z.B. die
individuelle Ernährungsform oder Alkoholkonsum eine Rolle spielen (Crawford et al. 1998, Powell 1994).
Zusammenfassend bestätigt sich der hohe Stellenwert der HFE-Gendiagnostik in Probanden mit V.a.
Eisenüberladung. Bei allen Probanden, die nicht homozygot für die C282Y-Mutation sind, sollte unbedingt
auch die H63D-Mutation getestet werden. Insgesamt zeichnet sich aber auch ab, dass die Gendiagnostik in
einigen Fällen nicht definitiv sein wird und allein die Diagnose einer hereditären Hämochromatose nicht
erbringen kann. Für die Diagnose und die Behandlungsnotwendigkeit ist auch weiterhin der Nachweis einer
klinisch relevanten Lebersiderose im Vergleich zum Lebensalter notwendig.
Literatur
Arnold C, Köck J, Weizsäcker Fv, Blum HE (1998) Hereditäre Hämochromatose. Deutsche med. Wochenschr 123: 397-398.
Borot N, Roth M, Malfroy M, Demangel C, Vinel JP, Pascal JP, Coppin H (1997) Mutations in the MHC class I-like candidate
gene for hemochromatosis in French patients. Immunogenetics 45:320-324.
Cardoso EMP, Stal P, Hagen K, Cabeda JM, Esin S, DeSousa M, Hultcrantz R (1998) HFE mutations in patients with
hereditary haemochromatosis in Sweden. J Int Med 243:203-208
Carella, M., D’Ambrosi, L., Totaro, A., Grifa, A., Valentino, M.A., Piperno, A., Girelli, D., et al (1997) Mutation analysis of the
HLA-H Gene in Italian Hemochromatosis patients Am J Hum Genet. 60: 828-832
Crawford DH, Jazwinska EC, Cullen LM, Powell LW (1998) Expression of HLA- linked hemochromatosis in subjects
homozygous or heterozygous for the C282Y mutation. Gastroenterol 114:1003-1008.
Cullen LM, Gao X, Easteal S, Jazwinska EC (1998) The hemochromatosis 845 G-->A and 187 C-->G mutations: prevalence in
non-Caucasian populations. Am J Hum Genet 62:1403-1407
Feder JN, Gnirke A, Thomas W, et al. (1996) A novel MHC class I-like gene is mutated in patients with hereditary
haemochromatosis. Nature Genetics 13:399-408
Feder JN, Tsuchihashi Z, Irrinki A, et al. (1997) The hemochromatosis founder mutation in HLA-H disrupts ß2-microglobulin
interaction and cell surface expression.
Feder JN, Penny DM, Irrinki A, et al. (1998) The hemochromatosis gene product complexes with the transferrin receptor and
lowers its affinity for ligand binding. Proceedings of the National Academy of Sciences of the United States of America
95:1472-1477.
Gottschalk R, Seidl C, Löffler T, Seifried E, Hoelzer D, Kaltwasser JP (1998) HFE codon 63/282 (H63D/C282Y) dimorphism in
German patients with genetic hemochromatosis. Tissue Antigens 51:270-275
Jazwinska EC, Cullen LM, Busfield F, Pyper WR, Webb SI, Powell LW, Morris CP, Walsh TP (1996) Haemochromatosis and
HLA-H. Nature Genetics 14:249-251
Jouanolle AM, Fergelot P, Gandon G, Yaouang J, LeGall JY, David V(1997) A candidate gene for hemochromatosis: frequency
of the C282Y and H63D mutations.
Lebron JA, Bennett MJ, Vaughn DE, et al. (1998) Crystal structure of the hemochromatosis protein HFE and characterization
of its interaction with transferrin receptor.
Mercier G, Bathelier C, Lucotte G (1998) Frequency of the C282Y mutation of hemochromatosis in five French populations.
Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ (1997) Global prevalence of putative haemochromatosis
mutations. Journal Med. Genet 34, 275-278
Messerschmitt C, Davion T, Capron JC et al. (1998) Phenotype/Genotype correlation in patients homozygous for the HFE
H63D mutation. Blood 92(10) Supplement 1(2):22b
Montalbetti N, Simonin A, Kovacs G, Hediger MA. (2013) Mammalian iron transporters: Families SLC 11 and SLC 40. Mol Asp
Med 34: 270-287 Nielsen P, Benn H-P, Peters C, et al. (1995) Iron status in prospective blood donors. Infusionsther
Transfusionsmed 22 (suppl): 142-144, 1995
Nielsen P, Fischer R, Engelhardt R, Dresow B, Gabbe EE (1998a) Neue Möglichkeiten in der Diagnose der hereditären
Hämochromatose. Deutsches Ärzteblatt 95:A2912-2921
Nielsen P, Carpinteiro S, Fischer R, Cabeda JM, Porto G, and Gabbe EE (1998b) Prevalence of the C282Y- and the H63D-
mutations in the HFE-gene in patients with hereditary haemochromatosis and in control subjects from Northern Germany.
Piperno A, Sampietro M, Pietrangelo A, et al. (1998) Heterogeneity of hemochromatosis in Italy. Gastroenterology 114:996-
1002.
Porto G, Alves H, Rodrigues P, et al. (1998) Major histocompatibility complex class I associations in iron overload: evidence
for a new link between HFE H63D mutation, HLA-A29, and non-classical form of hemochromatosis. Immunogenetics 47:404-
410.
Powell LW (1994) Primary iron overload. In: Brock JH, Halliday JW, Pippard MJ, Powell LW (Hrsg) Iron metabolism in health
and disease. Saunders, London, 227-270.
Rothenberg BE, Voland JR (1996) ß2m knockout mice develop parenchymal iron overload: A putative role for class 1 genes
of the major histocompatibility complex in iron metabolism. Proc Natl Acad Sci USA 93:1529-1534.
Santos M, Schilham MW , Rademakers LHPM, Marx JJM, De Sousa M, Clevers H (1996) Defective iron homeostasis in ß2-
microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med 184:1975-1985
Santos M, Clevers H, Marx JJM (1997) Mutations of the Hereditary hemochromatosis candidate gene HLA-H in Porphyria
Cutanea Tarda. N Eng J Med 336:1327-1328
Sham R, Ou C-Y, Braggins C, Phatak P (1998) Clinical characteristics of patients with hereditary hemochromatosis who are
H63D homozygotes.
Simon M, Alexandre JL, Bourel M, LeMarec B, Scordia C (1977) Heredity of idiopathic hemochromatosis: a study of 106
families. Clin Genet 11:327-341
Simon M, Alexandre JL, Fauchet R et al. (1980). The genetics of hemochromatose. In: Steinberg AG, Bearn AG, Motulsky et
al. Hrgb. Progrss in Medical Genetics. WB Saunders Philadelphia.
De Sousa M, Reimao R, Lacerda R, Hugo P, Kaufmann SEH, Porto G (1994) Iron overload in ß2-microglobulin-deficient mice.
Immunol Letters 39:105-111
Zhou XY, Tomatsu S, Fleming RE, et al. (1998) HFE gene knockout produces mouse model of hereditary hemochromatosis.
Proc Natl Acad Sci USA 95:2492-2497










© www.eiseninfo.de
Hämochromatose
Primäre, genetische Eisenüberladung,
hereditäre Hämochromatose
Der Begriff Hämochromatose geht auf von
Recklinghausen zurück, der fälschlicherweise
Hämoglobin als Ursprung der Eisenablagerung in der
Leber ansah. Unter diesem Begriff wurde anfangs eine
Eisenüberladung bei unterschiedlichen
Grunderkrankungen verstanden (3,4). Die heute
gängige Bezeichnung der hereditären (Synonym:
idiopathische, primäre) Hämochromatose wurde dann
einige Zeit allein für die HLA-assoziierte, genetisch
bedingte Form der Eisenüberladung verwendet (5)
(heute Typ 1 Hämochromatose).

Eisenüberladung
Aderlasstherapie. Methode aus dem Mittelalter!? Für Eisenspeicherkrankheit aber hochmodern