Heute kennen wir unter dem Begriff vier verschiedene genetische Formen der Hämochromatose (siehe Tab. 1), die
als Typ1 bis Typ 4 bezeichnet werden (6). Während Typ 1 bei uns häufig vorkommt, sind Typ 2-4 sind sehr seltene
Formen der erblichen Eisenspeicherkrankheit. Im Folgenden wird nur die HFE-assoziierte Hämochromatose
(Typ1) etwas näher beschrieben.
HFE-assoziierte Hämochromatose; Typ 1 Hämochromatose
Diese Form der hereditären Hämochromatose ist eine der häufigsten monogen vererbten Krankheiten der
kaukasischen Bevölkerung Nordeuropas, Amerikas, Australiens. Das HFE-Gen auf Chromosom 6 und die
ursächliche Mutation, die sogenannte C282Y-Mutation, wurden 1996 durch Feder et al. entdeckt. Die
Homozygoten-Frequenz ist ungefähr 1 auf 300 Personen in der Normalbevölkerung. Die Gen-Frequenz ist 1:20,
die Heterozygotenhäufigkeit ist 1 auf 10 Normalpersonen (6). Die Genhäufigkeit legt nahe, dass die heterozygote
Form einen Selektionsvorteil in der Evolution geboten hat. Heterozygote Genträger haben gegenüber der
Normbevölkerung eine leicht positive Eisenbilanz, was während ausgedehnter Hungerphasen, bei häufigen
Schwangerschaften oder für eine verbesserte Abwehr gegen Infektionen hilfreich gewesen sein kann. Nur die
C282Y-Mutation in homozygoter Form ist für die Hämochromatose Typ1 wichtig, alle anderen Varianten in dem
Gen sind klinisch eher bedeutungslos, von einigen unklaren Ausnahmefällen abgesehen.
Klinische Symptomatik
Bedingt durch die progressive Eiseneinlagerung in parenchymale Organe kann es meist im höheren Lebensalter
(ab 40-50 Jahren) zu vielfältigen klinischen Symptomen kommen (7). Die Leber-Eisenkonzentration ist ein guter
Anhalt für das Ausmaß der individuellen Eisenspeicherung. Es besteht eine Korrelation zwischen Leber-Eisen und
der Häufigkeit von Leberzirrhose, Diabetes und Hautpigmentierung. Dieser Zusammenhang ist nicht gegeben für
die Arthropathie, die vor allem die Metacarpophalangealgelenke der Finger betrifft und die in einigen Fällen erst
nach erfolgter Eisenentzugstherapie erstmals auftritt (8).
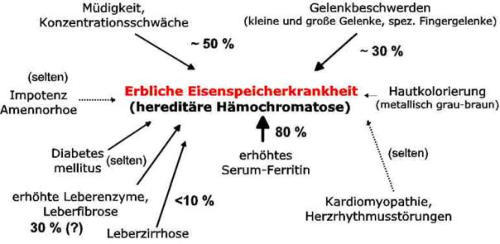
Abb. 1. Häufigkeiten von Symptomen und diagnostischen Merkmalen bei hered. Hämochromatose Typ1
(Zahlenangaben unveröffentlichte Ergebnisse)
Auffällig ist, dass die Häufigkeit von irreversiblen Schäden (Leberzirrhose, Diabetes) bei Diagnosestellung in den
letzten Jahren stark rückläufig ist. In unserem eigenen Patientenkollektiv, das in den letzten 8 Jahren diagnostiziert
worden ist, findet sich nur noch in ca. 10 % der Fälle eine Leberzirrhose. Dies ist zum einen auf eine verbesserte
Diagnostik und frühzeitige Therapie zurückzuführen, zum anderen scheint die Penetranz der HFE-assoziierten
Hämochromatose eher gering zu sein, sodass man heute auch viele klinisch wenig betroffene Patienten findet.
Zwei große Studien wiesen eine vergleichsweise niedrige klinische Ausprägung bei in Screeningstudien neu
entdeckten homozygoten C282Y-Trägern nach (9,10). Tab. 2:
Die C282Y-Homozygotie ist der einzig gängige Genotyp, der einen klinisch relevanten Hämochromatose-Phänotyp
produziert. Andere Mutations-Konstellationen (C282Y/H63D-Compound-Heterozygotie, H63D-Homozygotie),
führen nur in wenigen Einzelfällen zu signifikanten biochemischen oder klinischen Symptomen in Richtung
Eisenüberladung. Seit der Identifizierung des HFE-Gens 1996 hat es viele Untersuchungen zu möglichen
modifizierende Faktoren des phenotypischen Ausprägungsgrades der Hämochromatose gegeben. Dabei wurden
äußere Einflussfaktoren (Ernährung, Alkohol, Drogen, metabolisches Syndrom) genauso diskutiert wie genetische
Ursachen (häufige Polymorphismen in der BMP-Hepcidinaktivierungskaskade) (11). Aktuell werden auch
Variationen in der Hepcidin-Promotorregion diskutiert (12), die zu einem schweren Phänotyp mit besonders
niedrigen Hepcidinspiegeln führen (13).
Diese neueren Erkenntnisse sollten insgesamt keinesfalls dazu führen, die Typ1 Hämochromatose insgesamt zu
verharmlosen. Einzelne Patienten sind bereits in frühen Jahren schwer betroffen und weisen substantielle
eiseninduzierte Organschäden auf, sodass in jedem diagnostizierten Fall eine konsequente, vorsorgliche
Eisenentzugstherapie durch Aderlässe erfolgen sollte.
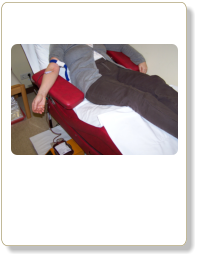
Die erschöpfende Aderlasstherapie (ca. 500 ml Blutentzug = 250 mg Eisen/Woche) ist nach wie vor die effektivste
Behandlungsmöglichkeit bei der erblichen Eisenspeicherkrankheit (14). Es wird solange therapiert, bis sich eine
leichte Eisenmangelanämie (Hb stabil < 12 g/dl, Serum-Ferritin < 30 µg/l) einstellt. Es folgt dann später eine
Erhaltungstherapie (3-6 Aderlässe/Jahr), die weiter überschüssig aufgenommenen Eisenmengen gleich entfernt
und somit einer Reakkumulation von Eisen dauerhaft entgegenwirkt.
Literatur
1.
Britton RS, Ferrali M, Magiera CJ, Recknagel RO, Bacon BR. Increased prooxidant action of hepatic cytosolic low-molecular-
weight iron in experimental iron overload. Hepatology 1990; 11:1038-1043
2.
Imlay JA, Chin SM, Linn S, Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science
1988; 240: 640-642
3.
von Recklinghausen FD. Tageblatt der 62. Versammlung Deutscher Naturforscher und Ärzte in Heidelberg, 1889; 324-325
4.
Sheldon JH. Haemochromatosis, Oxford University press, London 1935
5.
Simon M, Alexandre JL, Bourel M, LeMarec B, Scordia C. Heredity of idiopathic hemochromatosis: a study of 106 families.
Clin Genet 1977; 11:327-341.
6.
Pietrangelo A. Hereditary Hemochromatosis - A new look at an old disease. New Engl J Med 2004; 350:2383-2397
7.
Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk
VR, Leiendecker-Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P. Hemochromatosis and Iron
Overload Screening (HEIRS) Study: Screening of a primary care population. New Engl J Med 2005; 352:1769-1778
8.
Schumacher HR. Hemochromatosis and Arthritis. Arthritis Rheumatol 1964; 7:41-50
9.
Beutler E, Felitti VJ, Koziol JA, Ho JN, Gelbart T. Penetrance of 845G*A (C282Y) HFE hereditary haemochromatosis mutation
in the USA. Lancet 2002; 359:211-18
10.
Asberg A, Hveem K, Thorstensen K, Ellekjter E, Kannelonning K, Fjosne U, Halvorsen TB, Smethurst HB, Sagen E, Bjerve
KS : Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons.
Scand J Gastroenterol 2001; 36:1108-1115
11.
Deugnier Y, Mosser J. Modifying factors of the HFE hemochromatosis phenotype. Expert Rev Gastroenterol Hepatol. 2008
Aug;2(4):531-40.
12.
Island ML, Jouanolle AM, Mosser A, Deugnier Y, David V, Brissot P, Loréal O. A new mutation in the hepcidin promoter
impairs its BMP response and contributes to a severe phenotype in HFE related hemochromatosis. Haematologica. 2009 Mar
13. [Epub ahead of print] Links
13.
Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-
responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J
Mol Med DOI 10.1007/s00109-009-0447-2
14.
Powell LW. Primary iron overload. In: Brock JH, Halliday JW, Pippard MJ, Powell LW (Hrsg) Iron metabolism in health and
disease. Saunders, London 1994; 227-270
15.
Giardina PJ, Grady RW. Chelation therapy in betathalassemia: the benefits and limitations of desferrioxamine. Semin
Hematol 1995; 32: 304-312













© www.eiseninfo.de
Hämochromatose
Primäre, genetische Eisenüberladung,
hereditäre Hämochromatose
Der Begriff Hämochromatose geht auf von
Recklinghausen zurück, der fälschlicherweise
Hämoglobin als Ursprung der Eisenablagerung in der
Leber ansah. Unter diesem Begriff wurde anfangs
eine Eisenüberladung bei unterschiedlichen
Grunderkrankungen verstanden (3,4). Die heute
gängige Bezeichnung der hereditären (Synonym:
idiopathische, primäre) Hämochromatose wurde
dann einige Zeit allein für die HLA-assozierte,
genetisch bedingte Form der Eisenüberladung
verwendet (5).

Eisenüberladung
Aderlasstherapie. Methode aus dem Mittelalter!? Für Eisenspeicherkrankheit aber hochmodern