Veränderung
% der Patienten
Kopf (Mikro- o. Makrocephalus, flache Nase) 
21 %
Augen (Hypertelorismus, Epicanthus)
12 %
Nacken
4 %
Daumen (Hypoplasie, triphalangealer D.)
9 %
Nieren
7 %
Herz
7 %
Knochen
9 %
Andere
7 %
Wenigstens eine Veränderung
40 %
Tab. 1: Physische Veränderungen bei Kindern mit Diamond-Blackfan-Anämie
12-25 % der Patienten haben eine familiäre Geschichte von DBA. Der genaue Defekt im Knochenmark ist nicht
bekannt, er muss aber in der frühen Phase der Entwicklung der Roten Reihe liegen. Ca. 25 % der familiären
aber auch der sporadischen Fälle haben eine heterozygote Mutation im Gen (19q13.2) für das „ribosomal
protein 19“ (RPS19), das zur 40S-Ribosomenuntereinheit gehört (59). „Linkage“-Analysen von DBA-Familien
legen weitere veränderte Genorte nahe, z.B. 8p (8p23.3-p22) sowie 1q31 (58).
Die Behandlung der DBA besteht zunächst in einem Steroidtherapieversuch ab dem 2. Lebensjahr (initial
Prednison/Prednisolon 2 mg/kg/d, die eventuell notwendige Erhaltungsdosis individuell unterschiedlich, in der
Langzeitanwendung jedoch höchstens 0.5 mg/kg/d). In einer großen Studie an 222 Patienten aus Frankreich
und Deutschland zeigten 63 % der Patienten ein initiales Ansprechen mit Anstieg von Retikulozyten und
Erythrozyten nach 2-4 Wochen Steroidtherapie (61). In dieser Gruppe kam es bei 19 % der Patienten zu einer
Remission, die ohne weitere Therapie anhielt. 33 % blieben steroidabhängig, 10 % wurden resistent gegen
Prednisolon. Bei den 37 % der Patienten, die anfangs nicht auf Cortison ansprachen, bestand eine lebenslange
Transfusionspflichtigkeit. Nur in Einzelfällen wurde ein Ansprechen auf sehr hoch dosierte Steroidgaben (z.B.
Methylprednisolon-„Megadosen“) sowie Androgene, Metoclopramid, oder Interleukin-3 beobachtet. Bei
Patienten mit chronischer Transfusionspflichtigkeit sollte in der Familie nach einem HLA-kompatiblen
Stammzellspender gesucht werden, um ggf. möglichst frühzeitig (idealerweise im Alter von 2-3 Jahren) eine
Knochenmarkstammzelltransplantation durchführen zu können. Seit 1987 wurden in Deutschland 22
Knochenmarktransplantation bei DBA durchgeführt. Während eines medianen Beobachtungszeitraums von 5.9
Jahren (0.8-15.5 Jahren) überlebten 19 von 22 Patienten (62).
Bei DBA besteht im Rahmen der Grunderkrankung ein relativ hohes Risiko, im Erwachsenenalter eine maligne
Erkrankung zu entwickeln. Das kumulative Risiko für eine AML im Alter von 30-40 Jahren beträgt 23 % (63).
Auch solide Tumore kommen im Vergleich zur Normalbevölkerung gehäufter vor. Therapiebedingte
Komplikationen, die auch lebensbegrenzend sein können, ergeben sich durch die progressive Siderose bei
dauertransfundierten Patienten.
Abb. 1 zeigt die Lebereisenkonzentration bei einer Gruppe von DBA-Patienten aus Deutschland. Bei den
meisten Patienten zeigt sich eine substantielle Eisenüberladung der Leber. Auch die anderen typischen
siderosebedingten Komplikationen sind bei Patienten mit DBA unter regelmäßiger Transfusionstherapie zu
beobachten. So wurden wurde in einem europäischen Register bei 14% der Patienten Herzerkrankungen
beobachtet, bei bis zu 25% der Patienten treten endokrine Funktionsstörungen auf (7-25 % Hypogonadismus,
5% Diabetes mellitus) (61,63). Es sollte deshalb eine effektive Eiseneliminationstherapie angestrebt werden,
wobei eine evtl. myelotoxische Komponente des Eisenchelator speziell bei dieser Krankheit sehr kritisch
beachtet werden sollte. In Deutschland wurde deshalb bisher ausschließlich die Standard-Deferoxamin-
Therapie empfohlen (DFO subkutan über 10-12 Stunden: 40 -50 mg/kg/d an 5 Tagen/Woche). Studien mit dem
neuen oralen Chelatbildner Deferasirox, in die bisher u.a. mehr als 30 Patienten mit DBA eingeschlossen
wurden, zeigten eine gute Effektivität und Verträglichkeit dieses Medikamentes auch bei diesen Patienten
(zunächst über einen Auswertungszeitraum von 1 Jahr) ohne Hinweise auf eine eventuelle Myelotoxizität (64).
Die Zulassung von Deferasirox wird daher voraussichtlich auch die Behandlung von Patienten mit DBA
einschließen.
Abb. 1: Lebereisenkonzentration bei 37 Patienten mit DBA im Alter zwischen 2 und 33 Jahren, die dauerhaft
regelmäßig transfundiert werden. Eingezeichnet ist der für Thalassämie empfohlene Therapiebereich von 1.0-
2.1 mg/g (Lit 64). Viele Patienten mit DBA weisen eine deutlich höhere Lebereisenkonzentration auf.
Literatur
56. Chen S, Warszawski J, Bader-Meunier B, Tchernia G, Da-Costa L, Marie I, Dommergues JP. Diamond-
blackfan anemia and growth status: the French registry. J-Pediatr. 2005; 147(5):669-73
57. Gazda H, Lipton JM, Willig TN, Ball S, Niemeyer CM, Tchernia G, Mohandas N, Daly MJ, Ploszynska A,
Orfali KA, Vlachos A, Glader BE, Rokicka-Milewska R, Ohara A, Baker D, Pospisilova D, Webber A, Viskochil
DH, Nathan DG, Beggs AH, Sieff CA. Evidence for linkage of familial Diamond-Blackfan anemia to chromosome
8p23.3-p22 and for non-19q non-8p disease. Blood 2001; 97(7):2145-50
58. Da-Costa L, Tchernia G, Gascard P, Lo A, Meerpohl J, Niemeyer C, Chasis JA, Fixler J, Mohandas N.
Nucleolar localization of RPS19 protein in normal cells and mislocalization due to mutations in the nucleolar
localization signals in 2 Diamond-Blackfan anemia patients: potential insights into pathophysiology. Blood 2003;
101(12): 5039-45
59. Quigley JG, Gazda H, Yang Z, Ball S, Sieff CA, Abkowitz JL. Investigation of a putative role for FLVCR, a
cytoplasmic heme exporter, in Diamond-Blackfan anemia. Blood Cells Mol Dis 2005; 35(2):189-92
60. Willig TN, Niemeyer CM, Leblanc T, Tiemann C, Robert A, Budde J, Lambiliotte A, Kohne E, Souillet G,
Eber S, Stephan JL, Girot R, Bordigoni P, Cornu G, Blanche S, Guillard JM, Mohandas N, Tchernia.
Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-
Blackfan anemia patients. Pediatr Res 1999; 46(5):553-61
61. Niemeyer CM. Old and New Insights into Diamond Blackfan Anemia (DBA). Abstract. Symposium “Current
Issues in Iron Overload in rare anemias” Hamburg , 13-16 October, 2005, University Medical Center Hamburg -
Eppendorf
62. Janov AJ, Leong T, Nathan DG, Guinan EC. Diamond-Blackfan Anemia. Natural History and Sequelae of
Treatment. Medicine 1996; 75(2):77-87
63. Olivieri NF, Brittenham GM. Iron-Chelating Therapy and the Treatment of Thalassemia Blood 1997;
89:739-761
64. Greenberg PL, Young NS, Gattermann N. Myelodysplastic Syndromes. Hematology (Am Soc Hematol
Educ Program) 2002; 136-161












© www.eiseninfo.de
Diamond-Blackfan-Anämie
Die Diamond-Blackfan-Anämie (DBA) oder Blackfan-
Diamond-Anämie (BDA) ist eine seltene, kongenitale
hypoplastische Erythroblastopenie. 1 bis 7 Fälle treten bei
1,000,000 Geburten auf (55-58). Es gibt keine ethnischen
Unterschiede der Häufigkeit und beide Geschlechter sind
etwa gleich betroffen. Einige Kinder mit DBA (10-40 %)
zeigen physische Auffälligkeiten, wie z.B. einen verkürzten
oder auch einen triphalangealen (= Aase-Syndrom)-Daumen
(Tabelle 1). Viele Kinder sind klein, entwickeln sich aber
ansonsten normal. Der Defekt der Erythropoese wird meist
im Alter zwischen 3-12 Monaten erkennbar. Die Diagnose
wird anhand von Blut- und Knochenmarksausstrichen
gestellt, bei dem die normochrome, makrozytäre Anämie
sowie das Fehlen von erythropoetischen Vorläuferzellen bei
ansonsten normaler Zellentwicklung offensichtlich werden.
Andere bekannte Formen einer aplastischen Anämie müssen
ausgeschlossen werden. Die Adenosin-Deaminase-Aktivität
(eADA) in den Erythrozyten sowie der HbF-Anteil sind meist
gesteigert.

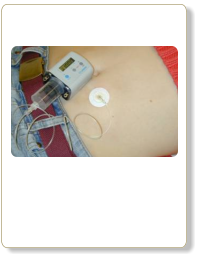
Eisenüberladung
Subkutane Injektion von DesferalR, einem Eisenchelator, der Eisen im Körper mobilisieren und ausscheiden lassen kann